
- SAM是一种序列比对格式标准, 由sanger制定,是以TAB为分割符的文本格式。主要应用于测序序列mapping到基因组上的结果表示,当然也可以表示任意的多重比对结果。
-
SAM要处理好的问题:
(1)非常多序列(read),mapping到多个参考基因组(reference)上;
(2)同一条序列,分多段(segment)比对到参考基因组上;
(3)无限量的,结构化信息表示,包括错配、删除、插入等比对信息;
-
SAM分为两部分,注释信息(header section)和比对结果部分(alignment section)。
-
注释信息可有可无,都是以@开头,用不同的tag表示不同的信息,主要有,说明符合标准的版本、比对序列的排列顺序;,参考序列说明;,比对上的序列(read)说明;,使用的程序说明;@CO,任意的说明信息。
-
比对结果部分,每一行表示一个片段的比对信息,包括11个必须的字段和一个可选字段。
第一列:QNAME,read name,read的名字通常包括测序平台等信息;
第二列:FLAG,sum of flags,每个数字代表一种比对情况,这里的值是符合情况的数字相加总和。
1 read是pair中的一条(read表示本条read,mate表示pair中的另一条read)
2 pair一正一负完美的比对上
4 这条read没有比对上
8 mate没有比对上
16 这条read反向比对
32 mate反向比对
64 这条read是read1
128 这条read是read2
256 第二次比对
512 比对质量不合格
1024 read是PCR或光学副本产生
2048 辅助比对结果
通过这个和可以直接推断出匹配的情况。假如说标记不是以上列举出的数字,比如说83=(64+16+2+1),就是这几种情况值和。
第三列:RNAME,reference sequence name,实际上就是比对到参考序列上的染色体号。若是无法比对,则是*;
第四列:POS,read比对到参考序列上,第一个碱基所在的位置,从1开始计数。若是无法比对,则是0;
第五列:MAPQ,Mapping quality,比对的质量分数,越高说明该read比对到参考基因组上的位置越唯一;
第六列:CIGAR,简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report),以参考序列为基础,使用数字加字母表示比对结果,比如3S6M1P1I4M,前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的;
“M”表示 match或 mismatch;
“I”表示 insert;
“D”表示 deletion;
“N”表示 skipped(跳过这段区域);
“S”表示 soft clipping(被剪切的序列存在于序列中);
“H”表示 hard clipping(被剪切的序列不存在于序列中);
“P”表示 padding;
“=”表示 match;
“X”表示 mismatch(错配,位置是一一对应的);
第七列:RNEXT,MRNM(chr),mate的reference sequence name,实际上就是mate比对到的染色体号,若是没有mate,则是*;
第八列:PNEXT,mate position,mate比对到参考序列上的第一个碱基位置,若无mate,则为0;
第九列:TLEN,Template的长度,ISIZE,Inferred fragment size.详见Illumina中paired end sequencing 和 mate pair sequencing,是负数,推测应该是两条read之间的间隔(待查证),若无mate则为0;
第十列:SEQ,Sequence,就是read的碱基序列,如果是比对到互补链上则是reverse completed eg.CGTTTCTGTGGGTGATGGGCCTGAGGGGCGTTCTCN
第十一列:QUAL,ASCII,read质量的ASCII编码。
第十二列之后:Optional fields,可选的区域
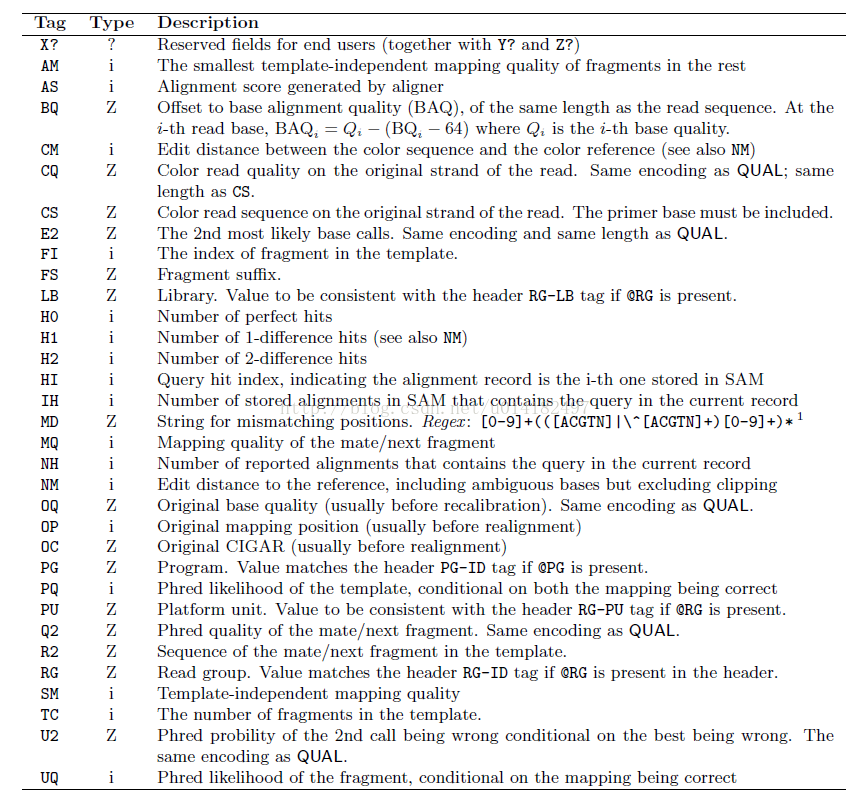
sam可选字段释义:
MC MC:Z:<CIGAR> 配对的那条read的CIGAR NM NM:i:<N> 插入/删除/替换的碱基总个数,不包含头尾被剪切的序列。一般来说等于序列中error base的个数 AS AS:i:<N> 匹配的得分。只有当匹配大于等于1出现。可为负,在local下为正。 XS XS:i:<N> 次优匹配的得分。当匹配大于1时出现。 XA XA:Z:<chr,pos,CIGAR,NM;chr,pos,CIGAR,NM;chr,pos,CIGAR,NM;...> 备选的。format: (chr,pos,CIGAR,NM;)* SA SA:Z:<chr,pos,+/-,CIGAR,MPQ,NM;> 第二最佳命中位置和标记(bwa的在vcf中表示为supplementary) MD MD:Z:<S> 比对上的错配碱基的字符串表示,不考虑软硬切 (替换:写reference原始碱基;插入:不表达;删除:^(删除的碱基)) RG RG:Z:<readID> read组ID